Étude Emergo : moins de demandes 510 (k) FDA pour les entreprises étasuniennes
有任何问题?向我们的专家获取相关信息
联系我们2017年 3月 23日
LES POINTS PRINCIPAUX PAR EMERGO :
- Les fabricants étasuniens de dispositifs médicaux soumettent moins de demandes d'autorisations 510 (k) que les années précédentes.
- Pour des questions liées, entre autres, aux devises, les soumissions de 510 (k) par des entreprises étrangères ont augmenté.
- Globalement, les chiffres des soumissions de 510 (k) ont chuté au cours des trois dernières années, alors que les délais de traitement des dossiers ont augmenté.
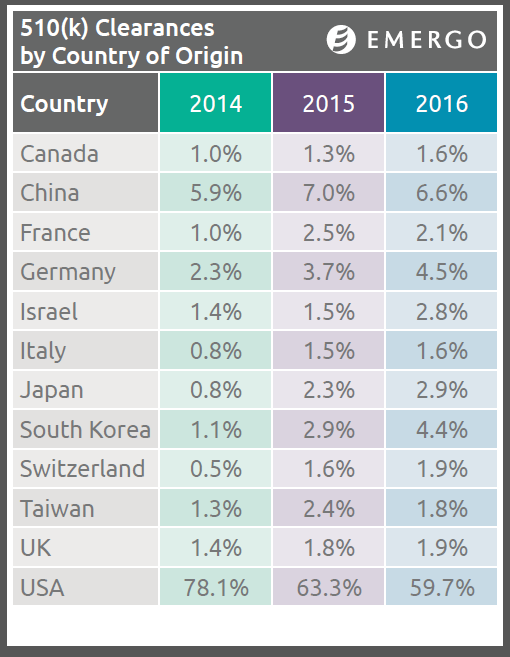 Selon une nouvelle étude d'Emergo, le nombre de fabricants de dispositifs médicaux basés aux États-Unis soumettant une notification précommercialisation 510 (k) à la Food and Drug Administration a diminué au cours des quatre dernières années, alors qu'il a dans le même temps augmenté pour ce qui est des entreprises asiatiques et européennes.
Selon une nouvelle étude d'Emergo, le nombre de fabricants de dispositifs médicaux basés aux États-Unis soumettant une notification précommercialisation 510 (k) à la Food and Drug Administration a diminué au cours des quatre dernières années, alors qu'il a dans le même temps augmenté pour ce qui est des entreprises asiatiques et européennes.
L'étude Emergo menée par Emergo s'est penchée sur environ 15 000 soumissions de 510 (k) autorisées par la FDA entre 2012 et 2016, indiquant non seulement une baisse régulière des demandeurs étasunien mais aussi que le total de 510 (k) acceptées par la FDA jusqu'en 2016 est en baisse sur une période de quatre ans. De nombreux facteurs aident à expliquer ces tendances —un dollar US (trop) fort, des pratiques d'examen plus strictes, des efforts vers l'exportation par les fabricants chinois et allemand—, mais ils méritent une analyse plus approfondie.
Les notifications 510 (k) étrangères dépassent celles des entreprises étasuniennes ?
L'une des constatations les plus frappantes de notre étude montre que les déclarations de notifications précommercialisation FDA des entreprises étasuniennes représentaient plus de 80 % du total en 2012 alors que ce chiffre tombe à moins de 60 % en 2016.
Lors de cette même période de 4 ans, les déclarations de firmes européennes et asiatiques —particulièrement les chinoises, allemandes et sud-coréennes, dont les pays ont des économies très portées vers l'exportation— ont quasi doublé, bien qu'en restant dans des pourcentages à 1 chiffre.
Les taux de change des devises ont joué un rôle important pour ces évolutions depuis 2012. Depuis 2014 le renforcement du dollar US a rendu l'importation de dispositifs médicaux moins couteuse pour les acheteurs étasuniens ; les fabricants étrangers ont réalisé que les taux de change favorables facilitent la compétition avec les entreprises étasuniennes sur le marché américain, et les chiffres du tableau ci-dessus illustrent cette tendance.
« Il est difficile de déterminer le pourquoi de la diminution du nombre d'entreprises étasuniennes présentant des 510 (k) » nous indique Chris Schorre, vice-président du marché international Emergo. « La FDA a certainement relevé la barre en ce qui concerne les exigences pour les tests et les données cliniques au cours des dernières années, et le coût associé d'une formalité d'autorisation 510 (k) aux États-Unis a augmenté. »
À titre d'exemple, C. Schorre cite des rapports indiquant que davantage de demandeurs 510 (k) sont invités à fournir des études d'utilisabilité sur leurs dispositifs, en particulier pour ceux en vente libre et utilisés par les consommateurs. Les tests de biocompatibilité ainsi que les demandes d'actions de vérification et de validation par les examinateurs de la FDA sont également devenus plus courants ; Les coûts associés ne sont pas négligeables.
« Bien que la FDA ne publie pas de détails sur la taille des entreprises qui soumettent des applications 510 (k), les jeunes et les petites entreprises obtiennent généralement l'approbation sur leur marché intérieur avant de s'étendre internationalement », explique C. Schorre. « Cela peut affecter de façon disproportionnée les petites entreprises étasuniennes de dispositifs médicaux qui peuvent ne pas être en mesure d'obtenir l'autorisation 501 (k) en raison du coût de la réponse aux exigences de la FDA. »
« Les entreprises étrangères présentant une demande 510(k) sont probablement mieux assises, avec plus de ressources financières et de personnel pour faire face à la charge accrue. »
Globalement moins de demandes 510 (k) et des temps de traitement plus longs
En lien avec le fait que moins d'entreprises étasuniennes demandent une autorisation 510 (k), cette dernière étude montre également une diminution du total de ces notifications précommercialisation 510 (k) auprès de la FDA.
L'année dernière, celle-ci a reçu le moins de demandes 510 (k) (soit 2 957) depuis 2010. Bien que le nombre des demandes provenant des entreprises asiatiques et européennes ait augmenté, ces augmentations ne peuvent se comparer à la baisse des demandes des entreprises américaines.
Outre les diminutions de demandes 510 (k), la FDA a également signalé des délais d'examen des demandes plus longs de 2011 à 2016 qu'à tout moment depuis le début jusqu'au milieu des années 1990. De plus, les temps d'examen moyens ont progressivement augmenté depuis 2001. Au cours de cette même période, les examinateurs de la FDA ont mis en œuvre des processus d'évaluation 510 (k) plus approfondis et plus stricts, ceci pouvant contribuer à expliquer ces délais accrus, même si l'ensemble du nombre de demandes diminue.
En 2016, le délai moyen d'examen pour un 510 (k) FDA étaient de 177 jours, en 2015 il était de 172 jours et il était de 178 jours en 2014. À nouveau, des exigences réglementaires plus strictes au cours des dernières années, concernant des points tels que les preuves cliniques et les tests, ont augmenté le temps nécessaire pour qu'un déclarant de notification avant la mise en marché obtienne d'être enregistré sur le marché étasunien.
« Alors que les orientations officielles de la FDA définissent comme objectif un examen des dossiers dans un délai de 90 jours, ne vous attendez pas à obtenir votre autorisation dans ce délai, » note le rapport, citant des données qui montrent que moins de 20 % de toutes les demandes 510 (k) présentées en 2016 ont été traitées en trois mois ou moins.
Quels types de DM sont les plus longs à être traités par la FDA ?
Enfin, la FDA a aussi fourni des statistiques sur la durée d'obtention du 510 (k) pour divers types de dispositifs médicaux. Alors que le délai moyen pour l'obtention de l'autorisation est de 177 jours, cette durée peut être légèrement moindre pour certains types de dispositifs, et beaucoup plus longue pour d'autres.
En 2016, les trois types de dispositifs pour lesquels la durée de traitement du dossier fut la plus longue étaient les produits d'immunologie (250 jours), produits d'hématologie (247 jours) et les produits d'anesthésiologie (245 jours). Ceux pour lesquels la durée fut la plus courte étaient les appareils de radiologie (112 jours), les produits de gastroentérologie et urologie (131 jours) et ceux de microbiologie (144 jours). De toute évidence, même pour les 510 (k) traités le plus rapidement, les autorités étasuniennes n'ont pas encore atteint les objectifs de leurs propres directives quant à un délai d'examen de 90 jours.
Les services et ressources d'Emergo en matière de 510 (k) FDA
- Consulting 510(k) pour les entreprises de dispositifs médicaux
- Calculateur interactif : combien de temps pour obtenir votre autorisation 510 (k) FDA ?
- Livre blanc : Testing requirements for 510(k) submissions to the FDA