Studie von Emergo: weniger 510(k)-Anträge von US-Unternehmen
有任何问题?向我们的专家获取相关信息
联系我们2017年 3月 17日
ZUSAMMENFASSUNG DER WESENTLICHEN PUNKTE DURCH EMERGO:
- US-Medizinprodukte-Hersteller stellen weniger 510(k)-Anträge als in der Vergangenheit.
- Die Anzahl der 510(k)-Anträge von ausländischen Herstellern hat sich aufgrund der Währungskurse und anderer Faktoren erhöht.
- Insgesamt hat die Anzahl der 501(k)-Anträge in den letzten drei Jahren abgenommen, wobei sich jedoch die Prüfzeiten erhöht haben.
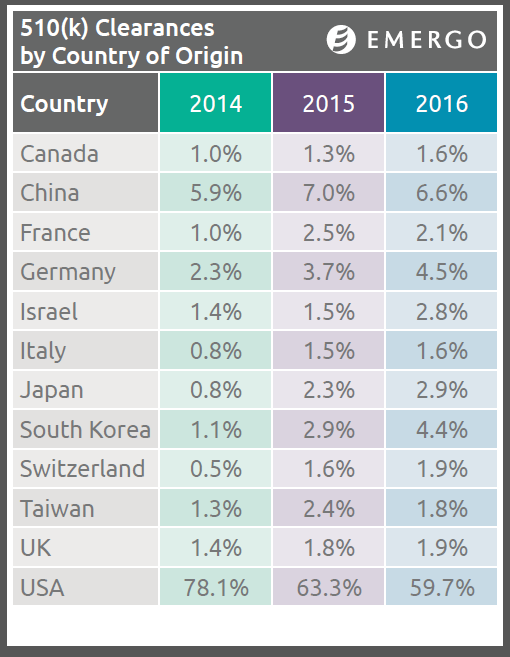
Die Anzahl der Medizinprodukte-Unternehmen mit Sitz in den USA, die bei der Food and Drug Administration 510(k)-Anträge gestellt haben, hat in den letzten vier Jahren abgenommen, wohingegen sich die Anzahl der Anträge von europäischen und asiatischen Firmen erhöht hat, wie eine neue Studie von Emergo ergeben hat.
In der jährlichen Studie hat Emergo ca. 15.000 510(k)-Anträge untersucht, die von der FDA zwischen 2012 und 2016 genehmigt wurden. Hierbei fanden wir nicht nur, dass die Anzahl der Antragsteller aus den USA stetig abgenommen hat, sondern auch, dass die Gesamtanzahl der 510(k)-Anträge, die von der Behörde genehmigt wurden, im Jahr 2016 ein Vierjahrestief erreicht hat. Die Gründe dafür sind vielfältig – ein (zu) starker US-Dollar, strengere FDA-Prüfungen, exportorientierte Bemühungen von chinesischen und deutschen Herstellern –, müssen jedoch tiefgehender analysiert werden.
Verdrängung von US-Unternehmen durch ausländische Hersteller?
Eines der verblüffendsten Ergebnisse unserer Studie war die Abnahme des Anteils der 510(k)-Anträge durch US-Unternehmen von mehr als 80 % aller solchen Anträge im Jahr 2012 auf unter 60 % im Jahr 2016.
Im selben Vierjahreszeitraum hat sich die Anzahl der Anträge von asiatischen und europäischen Firmen – insbesondere von Unternehmen mit Sitz in China, Deutschland und Südkorea mit stark exportorientierter Wirtschaft – ungefähr verdoppelt, obwohl die Anteile weiterhin im einstelligen Bereich bleiben.
Währungskurse spielten bei dieser Veränderung seit 2012 eine wesentliche Rolle. Seit 2014 sind importierte Medizinprodukte für US-Käufer aufgrund des starken US-Dollars kostengünstiger geworden. Ausländische Hersteller haben erkannt, dass die günstigen Wechselkurse den Wettbewerb mit US-Firmen in den USA erleichtern, was durch die Zahlen in der Tabelle oben bestätigt wird.
„Es ist schwer, zu orten, warum weniger US-Unternehmen 510(k)-Anträge einreichen“, sagt Chris Schorre, Vizepräsident für globales Marketing bei Emergo. „Die FDA hat sicherlich in den letzen Jahren die Latte in Bezug auf Studien und klinische Daten angehoben und die damit verbundenen Kosten für eine 510(k)-Freigabe in den USA sind angestiegen.“
Als Beispiel gibt Schorre Berichte an, die zeigen, dass von mehr 510(k)-Antragstellern Studien zur Gebrauchstauglichkeit für ihre Produkte angefordert werden, insbesondere für rezeptfrei erhältliche und von Endverbrauchern verwendete. Außerdem werden von den FDA-Prüfern häufiger Biokompatibilitätsprüfungen sowie Validierungen angefordert. Die damit verbundenen Kosten sind nicht unwesentlich.
„Obwohl die FDA keine Details zur Größe der Unternehmen freigibt, die 510(k)-Anträge einreichen, holen Start-ups und kleinere Unternehmen üblicherweise Freigaben in ihren Ursprungsmärkten ein, bevor sie international expandieren“, erläutert Schorre. „Dies kann sich unverhältnismäßig auf kleinere US-Medizinprodukte-Firmern auswirken, die aufgrund der Kosten, die mit der Erfüllung der FDA-Vorgaben verbunden sind, keine 510(k)-Freigabe erlangen können.
Ausländische Unternehmen, die 510(k)-Anträge bei der FDA einreichen, sind wahrscheinlich etablierter, mit größeren finanziellen und Personalressourcen, um die erhöhten Belastungen tragen zu können“, fährt Schorre fort.
Weniger 510(k)-Anträge insgesamt, längere Prüfzeiten
Abgesehen von der geringeren Anzahl der 510(k)-Anträge durch US-Hersteller zeigt die Studie auch, dass die Gesamtanzahl der Anträge bei der FDA sinkt.
Im Vorjahr erhielt die FDA die geringste Anzahl an 510(k)-Anträgen, 2.957, seit 2010. Obwohl die Anzahl der 510(k)-Anträge von europäischen und asiatischen Unternehmen angestiegen ist, kann diese Steigerung nicht mit der Abnahme der Anzahl der Anträge von US-Firmen mithalten.
Abgesehen von der Abnahme der 510(k)-Anträge meldete die FDA auch längere durchschnittliche Prüfzeiten von 2011 bis 2016 als in irgendeinem Zeitraum seit den frühen 90er-Jahren. Außerdem sind die durchschnittlichen Prüfzeiten seit 2001 stetig angestiegen. Im gleichen Zeitraum wurden gründlichere Prüfprozesse für 510(k)-Prozesse eingeführt, was die verlängerten Prüfzeiten sowie die Abnahme der Gesamtanzahl der Anträge erklären kann.
Im Jahr 2016 betrug die durchschnittliche 510(k)-Prüfzeit durch die FDA 177 Tage. Im Jahr 2015 war sie 172 Tage, im Vergleich zu 178 Tage im Jahr 2014. Hier wiederum trieben strengere regulatorische Anforderungen in Bezug auf klinische Nachweise und Studien die Prüfzeiten in die Höhe.
„Während die offiziellen FDA-Richtlinien das Ziel angeben, alle Anträge innerhalb von 90 Tagen zu prüfen, sollten Sie nicht erwarten, dass Ihr Antrag so schnell freigegeben wird“, so der Bericht mit der Angabe von Daten, nach denen weniger als 20 % aller 510(k)-Anträge, die im Jahr 2016 eingereicht wurden, innerhalb von drei Monaten oder darunter freigegeben wurden.
Bei welchen Medizinproduktetypen sind die Prüfzeiten am längsten?
Die FDA hat außerdem Daten dazu veröffentlicht, wie lange es dauert, bis 510(k)-Anträge für verschiedene Medizinproduktetypen genehmigt werden. 510(k)-Anträge werden durchschnittlich nach 177 Tagen genehmigt, bei bestimmten Produkttypen kann es jedoch etwas schneller gehen, bei anderen viel länger dauern.
Die drei Produkttypen, bei denen eine FDA-Freigabe im Jahr 2016 am längsten gedauert hat, sind Immunologieprodukte (250 Tage), Hämatologieprodukte (247 Tage) und Anästhesieprodukte (245 Tage). Produkttypen, bei denen die 510(k)-Prüfzeiten im Vorjahr am kürzesten waren, umfassen Radiologieprodukte (112 Tage), Gastroenterologie- und Urologieprodukte (131 Tage) und Mikrobiologieprodukte (144 Tage). Es ist klar, dass die FDA auch bei den kürzesten 510(k)-Prüfzeiten ihre eigene 90-Tage-Richtlinie verfehlt hat.
Beratungsleistungen und Ressourcen zu 510(k)-Anträgen von Emergo
- Beratung bei 510(k)-Anträgen für Medizinprodukte durch Emergo
- Interaktiver Rechner (auf Englisch): Wie lange wird die 510(k)-Freigabe für Ihr Produkt dauern?
- Whitepaper (auf Englisch): Prüfvorgaben für 510(k)-Anträge
作者
- Stewart Eisenhart