Emergo Study: FDA 510(k) Submissions from US Companies on the Decline
有任何问题?向我们的专家获取相关信息
联系我们2017年 3月 14日
EMERGO SUMMARY OF KEY POINTS:
- US medical device manufacturers are submitting 510(k) applications to the FDA less frequently than in previous years.
- Due to currency and other issues, 510(k) submissions from foreign companies have increased.
- Overall, numbers of 510(k) submissions have fallen over the past three years, while review times have increased.
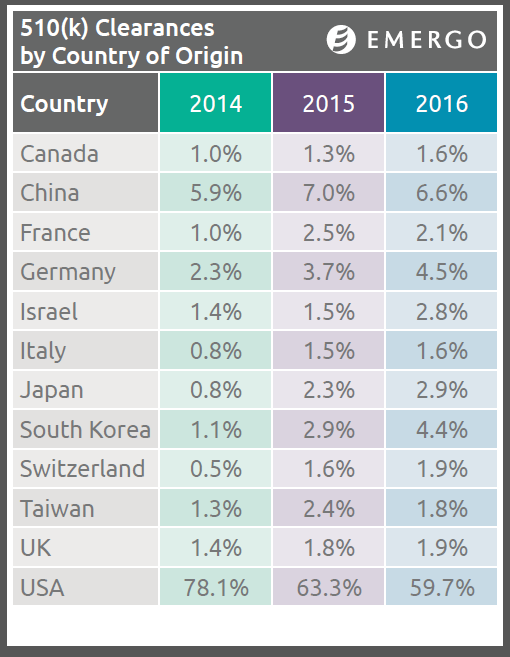 The number of US-based medical device manufacturers submitting 510(k) premarket notification applications to the Food and Drug Administration has decreased over the past four years, according to a new Emergo study, while applications from European and Asian firms have increased.
The number of US-based medical device manufacturers submitting 510(k) premarket notification applications to the Food and Drug Administration has decreased over the past four years, according to a new Emergo study, while applications from European and Asian firms have increased.
The annual Emergo study examined roughly 15,000 510(k) applications cleared by the FDA between 2012 and 2016, finding not only that numbers of US applicants has steadily declined, but also that the total number of 510(k) applications cleared by the agency hit a four-year low in 2016. Myriad factors—a (too) strong US dollar, more stringent FDA review practices, more export-oriented commercialization efforts by Chinese and German manufacturers—help explain these trends, but they deserve closer analysis.
Foreign 510(k) applicants squeezing out US firms?
One of the most striking findings in our study shows that FDA premarket notification submissions from firms in the US constituted more than 80% of all such submissions in 2012; in 2016, that percentage had fallen to just under 60%.
Over the same four-year period, submissions from Asian and European firms—particularly those in China, Germany and South Korea, all having aggressively export-oriented economies—roughly doubled, although they all remain in single-digit percentages.
Currency issues have played a significant role in how these percentages have changed since 2012. Since 2014, a strengthening US dollar has made imported medical devices less expensive for US purchasers; foreign manufacturers have realized that favorable exchange rates make it easier to compete with domestic firms in the US market, and the numbers in the chart above illustrate that trend.
“It’s hard to pinpoint why fewer US companies are submitting 510(k) applications,” says Chris Schorre, Vice President of Global Marketing at Emergo. “The FDA has certainly raised the bar with regard to requirements for testing and clinical data over the last few years, and the associated cost of getting 510(k) clearance in the US has risen.”
As an example, Schorre cites reports that more 510(k) applicants are being asked to provide usability studies on their devices, especially for those sold over the counter and used by consumers. Biocompatibilities testing as well as verification and validation activity requests by FDA reviewers have also become more common; associated costs are not insignificant.
“Although the FDA does not release details on the size of companies submitting 510(k) applications, startup and smaller firms typically get approval in their home market before expanding internationally,” Schorre explains. “This may be disproportionately affecting smaller US medical device companies that may not be able to pursue 501(k) clearance due to the cost of meeting FDA requirements.
“Foreign companies submitting 501(k) applications to the FDA are likely to be more established, with more financial and staff resources to accommodate the increased burden,” Schorre continues.
Fewer 510(k) applications overall, longer review times
Related to the findings that fewer US manufacturers are applying for 510(k) clearance, the latest study also indicates an overall decline in the number of premarket notification applications to the FDA.
Last year, the FDA received the fewest number of 510(k) applications—2,957—since 2010. Although 510(k) submissions from European and Asian companies have increased, these increases do not compare to the drop-off in applications from US firms.
Besides decreases in 510(k) submissions, the FDA has also reported longer average application review timeframes from 2011 to 2016 than at any point since the early to mid 1990s; furthermore, average review times have gradually increased since 2001. Over this same period, FDA reviewers have implemented more thorough and strict evaluation 510(k) processes, which may help explain these increased timeframes even as overall 510(k) submissions drop.
In 2016, average FDA 510(k) review times totaled 177 days; in 2015, it was 172 days, and in 2014 it was 178 days. Again, more stringent regulatory requirements regarding issues such as clinical evidence and testing in recent years has driven up the time it takes for a premarket notification applicant to obtain US market registration.
“While official FDA guidelines set a goal of reviewing all applications within 90 days, you should not expect your application to be cleared so swiftly,” the report notes, citing data that shows less than 20% of all 510(k) applications submitted in 2016 took three months or less to clear.
Which types of medical devices take longest to clear FDA review?
Finally, the FDA has also provided statistics on how long 510(k) applications for various types of medical devices take to clear the agency. While a 510(k) submission on average takes 177 days to clear, certain device types can take slightly less time; others can take much longer to undergo review.
The three types of devices that take the longest time to obtain FDA clearance in 2016 are immunology products (250 days), hematology products (247 days) and anesthesiology products (245 days). Device types with the shortest 501(k) review timeframes last year include radiology devices (112 days), gastroenterology and urology products (131 days) and microbiology products (144 days). Clearly even for 510(k)s with the shortest clearance timeframes, US regulators have yet to meet their own guidelines regarding 90-day review times.
Related FDA 510(k) services and resources from Emergo
- Emergo 510(k) application consulting for medical device companies
- Interactive calculator: How long will it take to clear your FDA 510(k) application?
- Whitepaper: Testing requirements for 510(k) submissions to the FDA