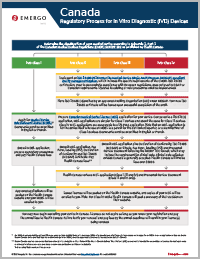
Health Canada Regulatory Approval Process for IVDs
有任何问题?向我们的专家获取相关信息
联系我们The chart shown illustrates the Health Canada approval process for In Vitro Devices (IVDs) and is available for download in PDF format. However, if you would like to explain the steps to someone else in an email, you can cut and paste the text below or send them a link to this page.
The Canadian approval process for IVDs explained
To obtain access to the Canadian market, in vitro diagnostic device manufacturers will need to secure a license. Health Canada issues two types of licenses: the Health Canada Medical Device Establishment License (MDEL) and the Health Canada Medical Device License (MDL). To see the difference between the two and find out more about the Health Canada approval process, please see or download the chart shown.
Step 1
Determine the classification of your medical device according to Schedule 1, Part 2 of the Canadian Medical Devices Regulations (CMDR) SOR/98-282 as published by Health Canada. IVDs fall into Class I, Class II, Class III or Class IV.
Step 2
For all devices except Class I, implement an ISO 13485:2016 under the Medical Device Single Audit Program (MDSAP) compliant quality management system, which includes the additional specific requirements of the CMDR. ISO 13485 certification, used to demonstrate compliance with European regulations, does not meet MDSAP or Canadian requirements. Updates to the existing or new procedures, must be implemented.
Step 3
For all devices except Class I, have ISO 13485 quality system (re)audited by an Auditing Organization (AO) under MDSAP. Several large European Notified Bodies also act as Registrars recognized by Health Canada. Your new ISO 13485 certificate will be issued upon successful completion of the audit.
Step 4
For Class I devices, apply for a Canadian Medical Device License (MDEL)* for your IVD. Documents must be submitted in English or French.
For Class II, III, and IV, Prepare Canadian Medical Device License (MDL) application for your device. Compared to a US 510(k) application, MDL applications are simpler for Class II devices and about the same for Class III devices. Class IV MDL applications are comparable to a US PMA application. Note that an MDL application is for the device itself whereas an MDEL is a permit for the distributor/importer, or a manufacturer of Class I devices. Documents must be submitted in English or French.
Step 5
For Class I, submit MDEL application, prepare mandatory procedures and pay Health Canada fees.
For Class II, submit MDL application, Fee Form, labeling (IFU), Declaration of Conformity and ISO 13485 (MDSAP) certificate. Pay Health Canada fees.**
For Classes III and IV, submit MDL application plus Declaration of Conformity, ISO 13485 (MDSAP) certificate, Fee Form, labeling (IFU) and Premarket Review Document following the IMDRF TOC format, which may include the requirement for clinical data. Clinical data collected outside Canada is generally accepted. Health Canada will invoice fees over $5000.
Step 6
Health Canada reviews MDL application (Class II, III and IV) and Premarket Review Document (Class III and IV only).
Step 7
For Class I, Approved applications will be posted on the Health Canada website and your MDEL certificate will be emailed to you.
For Classes II, III, and IV, issued licenses will be posted on the Health Canada website, and copies of your MDL will be emailed to you. Note: For Class IV MDLs, Health Canada will post a summary of their decision on their website.
Step 8
You may now begin marketing your device in Canada. Licenses do not expire as long as you renew your registration and pay the annual fees to Health Canada. Failure to file your renewal and pay fees by the annual deadlines will result in your license(s) being revoked.
* An MDEL is not required for retailers OR when using an importer/distributor with their own MDEL. If a manufacturer of a Class I device chooses not to apply for an MDEL, then no application or fees are due to Health Canada, but a qualified importer or distributor (with an MDEL) must be appointed before the device may be marketed in Canada.
** Health Canada requires a consumer field evaluation (clinical study) for all Near Patient IVDs. Health Canada defines “Near Patient” (IVD) as one that is intended for use outside a laboratory, for testing at home, or at the point of care, such as a pharmacy, a health care professional’s office or the bedside.
This is a simplified overview of the process.
Chart updated: 06/10/2019
相关
-
欧盟医疗器械法规MDR 全新解读 – 共瞻2022医疗器械合规新要点
自新欧盟医疗器械法规MDR (Medical Device Regulation 2017/745) 申请日期起,欧盟医疗器械监管当局就发布了一系列新文件及相关支持服务,旨在帮助医疗器械制造商适应新法规变更后的法规环境。 对此,作为在全球范围内经验丰富的医疗器械及IVD器械市场合
阅读更多 -
视频回顾 | 2022中美医疗健康产业投资研讨会
2022年11月23-25日,2022年度医疗器械行业盛会CMEF(中国国际医疗器械博览会)在深圳落下帷幕。展会期间,美国驻广州总领事馆、美国驻上海总领事馆携手国药励展于11月25日成功举办线上“中美医疗健康产业投资研讨会”,共享2022美国前沿医疗健康产业发展趋势及最新法规合规资讯。
阅读更多