马来西亚医疗器械注册和IVD器械认证合规审批流程
有任何问题?向我们的专家获取相关信息
联系我们此流程图阐释了马来西亚每个医疗器械分类的MDA注册审批流程,可在法规事务管理平台(RAMS)下载。
马来西亚医疗器械管理局(MDA)监管马来西亚的医疗器械和体外诊断(IVD)器械。所有器械都需要上市前审批,进入马来西亚市场的制造商将能利用他们在公认参考市场的现有批准来简化审查流程。
马来西亚医疗器械管理局(MDA)医疗器械审批流程详解
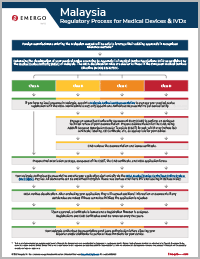
此流程图阐释了马来西亚每个医疗器械分类的MDA审批流程,可在法规事务管理平台(RAMS)下载。这里简要概述了注册流程的主要步骤。
第1步
根据MDA的分类规则判定您的器械的分类。
第2步
任命一名当地授权代表(AR)管理您的医疗器械注册,并代表您与MDA互动。
第3步
编制ASEAN通用递交档案模板(CSDT)并聘请一个认可的符合性评估机构(CAB)来审查您的文件。低风险器械免于CAB审查。
第4步
CAB审查您的文件,如果通过评估,将会签发符合性评估报告和证书。
第5步
编制所需的所有文件、表格和CBA证书(如果适用)以便递交给MDA。
第6步
您的AR通过MDA的线上系统以电子方式递交申请,并支付申请费用。
第7步
MDA将审查申请,并可能要求补充信息。
第8步
批准后需要支付注册费。支付后,MDA签发载有注册号的注册证书。
相关
-
韩国医疗器械法规框架及注册路径解读
我们的专家将详细解读2023年韩国食品药品安全部(MFDS)指导文件草案更新下韩国医疗器械注册监管要求、分类标准、韩国良好生产规范(KGMP)等相关要求,并将依据多年韩国本地注册经验提供注册案例分享。此外,您还将了解到制造商在面对MFDS监管时需要关注的核心要点,相关研讨会内容包括:
阅读更多 -
美国医疗器械咨询服务
阅读更多