Rapport de la FDA des États-Unis : améliorations liées à la RTA et délais d'examen réduits
有任何问题?向我们的专家获取相关信息
联系我们2015年 8月 31日
Un nouveau rapport produit par la FDA des États-Unis indique une réduction sensible des cas où des dossiers avant commercialisation incomplets sont refusés par la RTA policy, et une diminution générale des délais d'examen pour les autorisations et homologations entre les exercices financiers 2010 à 2014.
Le rapport en anglais indique des délais pour les notifications avant mise en marché 510 (k), les autorisations pré-commercialisation ou APMM, les exemptions pour dispositif expérimental (IDE) et les évaluations de novo de nouvelles technologies et nouveaux dispositifs, en citant diverses modifications de programmes et de politiques internes depuis 2010, ainsi que des facteurs externes liés aux lois Medical Device User Fee Act (MDUFA — Loi sur les frais d'utilisation des dispositifs médicaux) et Food and Drug Administration Safety and Innovation Act (FDASIA— Loi sur la sécurité et l'innovation FDA).
Moins de refus RTA en 2015
L'autre indication importante du rapport concerne la stratégie RTA (Refuse to Accept) par laquelle les dossiers des demandeurs de 510 (k) sont pré-examinés et refusés s'il leur manque des éléments requis.
La FDA indique une diminution de 20 % de ces cas : le taux de refus RTA tombe de 57 % au premier trimestre 2014 à 37 % au deuxième trimestre 2015. La FDA attribue cette diminution à une habitude mieux diffusée, une stratégie RTA claire pour l'industrie, qui ont donc mené à moins d'erreurs et d'oublis dans les dossiers 510 (k) présentés. Depuis la mise en œuvre de la stratégie RTA au début de l'année 2013, le taux de refus a diminué de façon constante, ce qui suggère une meilleure connaissance des éléments nécessaires à un dossier 510 (k) de notification préalable à la commercialisation, pour se diriger vers un examen approfondi de la FDA.
Des réductions substantielles pour les IDE, évaluations de novo
Les réductions les plus spectaculaires ont été signalées pour les évaluations de novo et les Exemptions pour dispositif expérimental (IDE) ; selon la FDA, les délais d'examen IDE ont diminués de plus d'un an et les évaluations de novo sont passées de plus de trois ans en 2010 à 10 mois en 2014. Comme l'indique la figure 1, les délais d'examen de 510 (k) et APMM ont respectivement diminués de 13 % et 31 %.
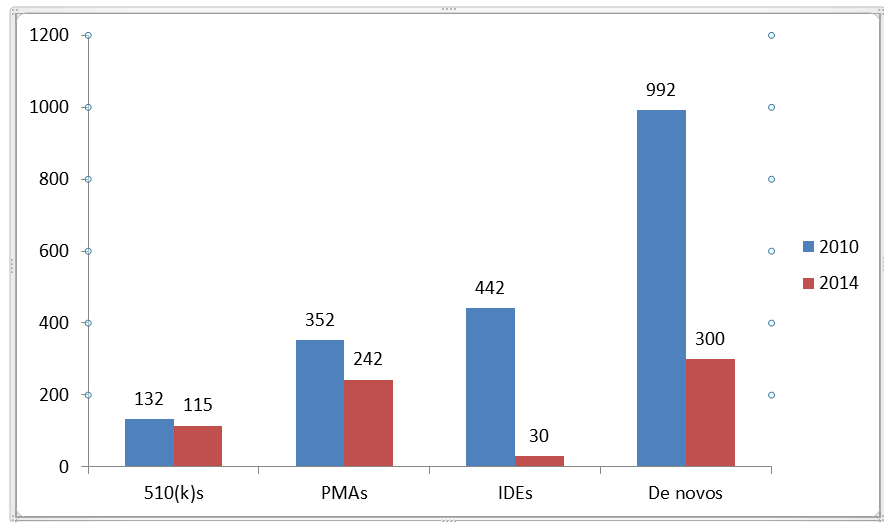
Figure 1 : Délais d'examen en nombre de jours pour FYE 2010 et 2014 (source: US FDA)
En ce qui concerne les délais très raccourcis des examens de novo, ceci peut peut-être s'expliquer par l'autorisation donnée par la FDA aux fabricants de transmettre leur demande pour dispositif de novo sans passer directement d'abord par un examen 510 (k) et l'obtention de décisions de non-équivalence substantielle (Not Substantially Equivalent — NSE).
Des délais d'examen plus courts signifient davantage d'autorisations, d'homologations
Le rapport de la FDA montre également comment la diminution des délais a conduit à plus d'homologations ou autorisations de commercialisation sur le marché américain.
Il est précisément indiqué que le nombre de demandes 510 (k) acceptées avait atteint 84 % en 2014 alors qu'il était à 73 % en 2010. Les APMM acceptées par la FDA sont également à 86 % en 2014 contre 59 % en 2010.
Points clés
- Des diminutions visibles des refus RTA sont dues à la sensibilisation de la FDA, ainsi qu'une plus grande connaissance par le secteur des exigences liées aux demandes 510 (k)
- Un processus de demande de novo plus direct a réduit les délais d'autorisation de mise sur le marché pour ces types de dispositifs médicaux
- Des modifications de programme et de stratégie au CDRH (Center for Devices and Radiological Health) ont abouti à des délais d'examen diminués pour les demandes 510(k) et APMM